

El diagnóstico de la enfermedad de Parkinson (EP) sigue siendo clínico, pero en estos últimos años se han producido importantes avances. En este capítulo revisamos los criterios diagnósticos vigentes, el papel actual de los marcadores sugestivos de sinucleinopatía subyacente con más rendimiento diagnóstico, la utilidad de los test farmacológicos y el diagnóstico diferencial.
El diagnóstico correcto de la EP es fundamental para el manejo óptimo del paciente, pero obtener una certeza diagnóstica completa en vida sigue siendo hoy en día imposible, a pesar de décadas de avances. En los estudios clinicopatológicos realizados en la década de los noventa se consiguió una confirmación diagnóstica por autopsia de entre el 75 y el 95% en los pacientes que habían sido diagnosticados de EP por expertos1-4 (NE-II).
La precisión diagnóstica varía considerablemente según la duración de la enfermedad, la edad, la experiencia del médico y la evolución en nuestra comprensión de la EP (estudios más recientes muestran en general una mayor precisión).
Los errores en el diagnóstico pueden ser atribuibles a la falta de reconocimiento de otras patologías neurodegenerativas, causas de parkinsonismo secundario o de la ausencia de un auténtico parkinsonismo (por ejemplo, temblor esencial, temblor distónico, etc.). Desde la descripción de la EP se han producido cambios muy importantes tanto en aspectos clínicos, etiofisiopatológicos y terapéuticos como en el concepto que tenemos sobre la enfermedad. Entendemos mejor las manifestaciones motoras y el papel de su respuesta al tratamiento como parte de los criterios diagnósticos. Tenemos una mayor claridad en las definiciones anatomopatológicas, entendiendo que la neurodegeneración comienza antes de que se manifiesten los síntomas motores, y un mayor conocimiento sobre las manifestaciones no motoras y sobre el papel que tienen los factores genéticos y epigenéticos en la etiofisiopatología de la enfermedad.
También en los últimos años se han hecho importantes avances en el campo de los biomarcadores. Así, en el ámbito de los biomarcadores de laboratorio, hemos vivido el desarrollo de ensayos de amplificación de semilla (en inglés seeding amplification assays [SAA]), que permiten evidenciar in vitro la capacidad proagregante de α-sinucleína en varias muestras biológicas, con sensibilidad y especificidad óptimas por ahora en el líquido cefalorraquídeo (LCR), si bien con resultados prometedores en otros fluidos y tejidos (sangre, piel, etc.)5. Sin embargo, los biomarcadores de imagen presentan limitaciones. La imagen estructural por resonancia magnética (RM) (con signos como el swallow tail sign, por alteración del nigrosoma, o las secuencias de neuromelanina) aún no ha sido evaluada apropiadamente en términos de especificidad comparada con otros parkinsonismos, mientras que la imagen molecular de α-sinucleína mediante pruebas de medicina nuclear (fundamentalmente la tomografía por emisión de positrones [PET]), lleva años encontrando dificultades mayores a las observadas con la imagen molecular de amiloide-β y τ, y, por ahora, la escasa literatura científica ha mostrado algunos resultados más sólidos en atrofia multisistémica que en la propia EP6-8.
Estos avances en el conocimiento nos han obligado a replantearnos aspectos básicos, como son la definición de la EP y sus criterios diagnósticos.
De este modo, en 2015, el grupo de trabajo de la Sociedad Internacional de la Enfermedad de Parkinson y Trastornos del Movimiento (Movement Disorder Society [MDS]) presentó los criterios diagnósticos de investigación para la fase prodrómica9 (NE-IV) y para uso en la clínica para la EP clínicamente manifiesta10 (NE-IV). Estos criterios surgieron del trabajo de un grupo de expertos internacionales en el que revisaban la definición de la EP y algunos aspectos críticos que comentaremos a continuación11 (NE-IV). En primer lugar, se replanteó el papel central de la patología clásica como algo indispensable para el diagnóstico, debido a la existencia de nueva evidencia sobre EP de causa genética sin depósito de sinucleína, una alta prevalencia de cuerpos de Lewy incidentales y que los agregados de α-sinucleína pueden no estar presentes en fases preclínicas de la enfermedad12-15. Igualmente, la identificación de formas genéticas y familiares de la enfermedad condujeron a eliminar la historia familiar como criterio de exclusión. En segundo lugar, se reconoció la demencia como un aspecto frecuente en la EP, proponiendo la consideración de la demencia por cuerpos de Lewy (DCL) como una variante fenotípica de la EP y eliminando, por tanto, la presencia de demencia como un criterio de exclusión. Por último, se hizo énfasis en la importancia de reconocer la heterogeneidad de la enfermedad y la existencia de una fase prodrómica cuya caracterización permitiría el uso de estrategias neuroprotectoras cuando estuvieran disponibles.
Estos criterios fueron desarrollados para la investigación en este campo y hoy en día aún no se contempla su uso en la práctica clínica diaria. La EP prodrómica se define como la fase de la enfermedad en la que los síntomas secundarios a la neurodegeneración están presentes, pero el diagnóstico clínico de la EP no es todavía posible. Los criterios se basan en un modelo que calcula la probabilidad de sufrir una EP prodrómica tomando como base la edad y añadiendo información sobre factores de riesgo ambientales y genéticos, así como el resultado de pruebas complementarias (PET-tomografía por emisión de fotón único [SPECT], ecografía cerebral, etc.)16-18 (NE-III). Estos criterios han sido validados en cohortes prospectivas (NE-II) y tienen una especificidad relativamente alta para la conversión de una EP prodrómica probable (> 80% de probabilidad) a una EP clínica. En cambio, su sensibilidad es variable, porque depende de los marcadores presentes (se requieren marcadores altamente predictivos para una alta sensibilidad) y del tiempo de evolución del proceso patológico asociado a la EP (la sensibilidad disminuye en puntos temporales tempranos, cuando muchas características no han tenido tiempo de desarrollarse)19 (NE-II). En 2019 se publicó una actualización de estos criterios20 (NE-IV), añadiendo nuevos marcadores prodrómicos y revisando los valores predictivos de los previamente incluidos. Estos criterios actualizados presentaron una sensibilidad ligeramente mejor que los originales, pero en general por debajo del 65%21-24 (NE-II). Utilizando un punto de corte del 80%, la sensibilidad fue del 0% (en la cohorte Hellenic Longitudinal Investigation of Aging and Diet [HELIAD]), del 35% (la cohorte Tübingen Evaluation of Risk Factors for Early Detection of Neurodegeneration [TREND]) con 10 años de seguimiento22, del 40% (en el estudio Bruneck25 a 10 años de seguimiento23) y del 64% (en el estudio Bruneck25 a 5 años de seguimiento23) y utilizando un punto de corte más bajo del 50%, la sensibilidad aumentó al 4,5% (HELIAD), el 65% (TREND: 10 años de seguimiento, Bruneck: 10 años de seguimiento) y el 91% (Bruneck: 5 años de seguimiento)22-25.
Estos criterios fueron creados fundamentalmente para su uso en investigación, pero tras haber sido validados frente al gold standard26 (el diagnóstico clínico realizado por un experto y con casos confirmados neuropatológicamente)27, la MDS respalda su uso en la práctica clínica diaria28 (NE-II). Los aspectos motores y la mayoría de las características diagnósticas se identifican a través de una cuidadosa historia clínica y un examen neurológico detallado. Por consiguiente, el diagnóstico exige una formación adecuada en Neurología.
Primero se evalúan las alteraciones motoras (definidas como bradicinesia más temblor de reposo o rigidez), consideradas el eje central del diagnóstico de parkinsonismo. El siguiente paso se basa en confirmar la existencia o ausencia de criterios de exclusión y de banderas rojas, además de confirmar la existencia de otros criterios positivos adicionales que sirven de apoyo y aumenten la confianza a la hora de realizar el diagnóstico.
Se incluyen dos niveles distintos de certeza:
Otras características clave para la valoración de los criterios diagnósticos son las siguientes:
El parkinsonismo se define como bradicinesia, en combinación con temblor de reposo, rigidez o ambos. Estos hallazgos deben ser claramente demostrables y no atribuibles a factores de confusión (envejecimiento, artritis, debilidad, etc.). El examen de todos los signos cardinales debe realizarse tal y como se describe en la sección de exploración motora (parte III) de la Escala Unificada de la Enfermedad de Parkinson Modificada por la Sociedad de Trastornos del Movimiento (MDS-UPDRS):
Aunque la inestabilidad postural es una característica del parkinsonismo, no se considera parte de los criterios MDS-PD. Se presenta de forma frecuente en estadios tardíos de la EP, pero su presencia en estadios precoces sugiere un diagnóstico alternativo.
Una vez establecido el diagnóstico de parkinsonismo se aplican los criterios MDS-PD para determinar si la EP es la causa del parkinsonismo.
El diagnóstico de EP clínicamente establecida requiere:
El diagnóstico de EP clínicamente probable puede hacerse si:
Una respuesta clara al tratamiento dopaminérgico. Para cumplir este criterio, el paciente, durante su tratamiento inicial, debe regresar casi hasta la situación funcional normal previa. No es suficiente que se documente “algo de mejoría”. La respuesta debe ser inequívoca y En ausencia de documentación de esta respuesta inicial (poca información, uso de agentes con menor eficacia o dosis bajas), la mejoría necesaria para cumplir el criterio se puede definir como una marcada mejoría a medida que se aumenta la dosis o un marcado empeoramiento con la reducción de la dosis. Se puede documentar de forma objetiva con 1) un cambio de más del 30% en la escala MDS-UPDRS parte III tras el cambio de tratamiento, o 2) la aparición inequívoca y marcada de fluctuaciones.
Para todos los criterios de exclusión y para las banderas rojas se asume que, si el criterio no se cumple por una causa alternativa y no relacionada, no constituye un criterio de exclusión (por ejemplo, si se observan anormalidades cerebelosas unilaterales atribuibles a un ictus cerebeloso).
La presencia de cualquiera de los siguientes hallazgos descarta una EP:
Se consideran banderas rojas las siguientes:
La disfunción autonómica es un hallazgo frecuente en la EP. Este criterio pretende identificar la disfunción autonómica grave asociada particularmente a la AMS.


Rajput AH, Rozdilsky B, Rajput Accuracy of clinical diagnosis in parkinsonism: a prospective study. Can J Neurol Sci. 1991;18:275-8.
Hughes AJ, Daniel SE, Lees Improved accuracy of clinical diagnosis of Lewy body Parkinson’s disease. Neurology. 2001;57:1497-9.
Hughes AJ, Daniel SE, Kilford L, et Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55:181-4. d
Tolosa E, Wenning G, Poewe W. The diagnosis of Parkinson’s disease. Lancet Neurol. 2006;5:75-86.
Bellomo G, De Luca CMG, Paoletti FP, et al. α-Synuclein Seed Amplification Assays for Diagnosing Synucleinopathies: The Way Forward. Neurology. 2022;99(5):195-205.
Mahlknecht P, Krismer F, Poewe W, et Meta-analysis of dorsolateral nigral hyperintensity on magnetic resonance imaging as a marker for Parkinson›s disease. Mov Disord. 2017;32(4):619-23.
Chougar L, Arsovic E, Gaurav R, et al. Regional Selectivity of Neuromelanin Changes in the Substantia Nigra in Atypical Parkinsonism. Mov Disord. 2022;37(6):1245-55.
Smith R, Capotosti F, Schain M, et al. The α-synuclein PET tracer [18F] ACI-12589 distinguishes multiple system atrophy from other neurodegenerative Nat Commun. 2023;14(1):6750.
Berg D, Postuma RB, Adler CH, et al. MDS research criteria for prodromal Parkinson›s Mov Disord. 2015;30(12):1600-11.
Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson›s disease. Mov Disord. 2015;30(12):1591-601.
Berg D, Postuma RB, Bloem B, et Time to redefine PD? Introductory statement of the MDS Task Force on the definition of Parkinson›s disease. Mov Disord. 2014;29(4):454-62.
Kalia LV, Lang AE, Hazrati LN, et al. Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol. 2015;72(1):100-5.
Siderowf A, Concha-Marambio L, Lafontant DE, et al. Assessment of heterogeneity among participants in the Parkinson’s progression markers initiative cohort using alpha-synuclein seed amplification: a cross-sectional Lancet Neurol. 2023;22(5):407-17.
Van de Berg WD, Hepp DH, Dijkstra AA, et al. Patterns of α-synuclein pathology in incidental cases and clinical subtypes of Parkinson›s disease. Parkinsonism Relat Disord. 2012;18 Suppl 1:S28-30.
Chahine LM, Merchant K, Siderowf A, et al. Proposal for a Biologic Staging System of Parkinson›s Disease. J Parkinsons Dis. 2023;13(3):297-309.
Mahlknecht P, Gasperi A, Willeit P, et al. Prodromal Parkinson›s disease as defined per MDS research criteria in the general elderly Mov Disord. 2016;31(9):1405-8.
Fereshtehnejad SM, Montplaisir JY, Pelletier A, et al. Validation of the MDS research criteria for prodromal Parkinson’s disease: longitudinal assessment in a REM sleep behavior disorder (RBD) cohort. Mov Disord. 2017;32:865-73.
Mirelman A, Saunders-Pullman R, Alcalay RN, et al.; AJ LRRK2 Consortium. Application of the Movement Disorder Society prodromal criteria in healthy G2019S-LRRK2 carriers. Mov Disord. 2018;33(6):966-973.
Pilotto A, Heinzel S, Suenkel U, et Application of the movement disorder society prodromal Parkinson›s disease research criteria in 2 independent prospective cohorts. Mov Disord. 2017;32(7):1025-1034.
Heinzel S, Berg D, Gasser T, et al.; MDS Task Force on the Definition of Parkinson›s Disease. Update of the MDS research criteria for prodromal Parkinson›s Mov Disord. 2019;34(10):1464-1470.
Giagkou N, Maraki MI, Yannakoulia M, et al. A Prospective Validation of the Updated Movement Disorders Society Research Criteria for Prodromal Parkinson›s Disease. Mov 2020;35(10):1802-1809.
Yilmaz R, Suenkel U, TREND Study Team; et Comparing the Two Prodromal Parkinson›s Disease Research Criteria-Lessons for Future Studies. Mov Disord. 2021;36(7):1731-1732.
Marini K, Mahlknecht P, Tutzer F, et Application of a Simple Parkinson›s Disease Risk Score in a Longitudinal Population-Based Cohort. Mov Disord. 2020;35(9):1658-1662.
Giagkou N, Maraki MI, Yannakoulia M, et A prospective validation of the updated movement disorders society research criteria for prodromal Parkinson’s disease. Mov Disord. 2020;35(10):1802-1809.
Kiechl S, Willeit J. In a nutshell: findings from the Bruneck study. Gerontology. 2019;65(1):9-19.
Postuma RB, Poewe W, Litvan I, et al. Validation of the MDS clinical diagnostic criteria for Parkinson›s disease. Mov Disord. 2018;33(10):1601-1608.
Virameteekul S, Revesz T, Jaunmuktane Z, et al. Clinical Diagnostic Accuracy of Parkinson›s Disease: Where Do We Stand? Mov Disord. 2023;38(4):558-566.
Diagnosis of Parkinson’s disease. En: International Parkinson and Movement Disorder Society [Internet]. Disponible en: https://ur0.jp/H8XpR
McKeith IG, Dickson DW, Lowe J, et al; Consortium on DLB. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65(12):1863-72.
Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(Pt 9):2456-77.
Para la aceptación de un biomarcador, la sensibilidad y la especificidad, en particular, tienen que ser elevadas (superiores al 80%). Actualmente, entre los marcadores de α-sinucleinopatía únicamente cumplirían estas condiciones los de amplificación de α-sinucleína (αSyn-SAA, por sus siglas en inglés) en LCR y piel de forma inconsistente en la literatura científica31-33 (NE-II). El uso de otras técnicas (por ejemplo, ELISA o SIMOA para medir los niveles de α-sinucleína (o de las técnicas mencionadas previamente), pero utilizando otros tejidos, estarían reservadas para el campo de la investigación en el momento actual, por lo que no se comentarán en este capítulo.
Los SAA son técnicas novedosas y ultrasensibles que permiten detectar la capacidad proagregante de una proteína incluso en muestras con cantidades ínfimas de proteínas mal plegadas. Estas técnicas se desarrollaron inicialmente para el estudio de las encefalopatías espongiformes transmisibles usando la amplificación cíclica de mal plegamiento de proteínas (del inglés, protein misfolding cyclic amplification [PMCA]) para evaluar la presencia de proteínas priónicas patológicas. Posteriormente, reemplazando la sonificación por agitación vigorosa y combinando ensayos de fluorescencia de tioflavina T, resultó la conversión inducida por agitación en tiempo real (del inglés, real time quaking-induced conversion [RT-QuIC]), técnica más utilizada hoy en día en estas enfermedades, aunque el término genérico de SAA se está empezando a imponer.
Durante la última década, haciendo uso de la propagación interneuronal de la α-sinucleína, similar al de las enfermedades por priones, los SAA se han extendido en el campo de las sinucleinopatías (EP idiopática con y sin demencia, con resultados variables en la EP genética y la AMS)31,34 (NE-II). Una revisión sistemática y metaanálisis reciente36 (NE-I) que incluye 36 estudios con 2722 pacientes con sinucleinopatías (algunos diag- nosticados clínicamente y otros patológicamente) y 2278 controles, demostraron una sensibilidad y especificidad agrupadas de los SAA de α-sinucleína de 0,91 (0,87-0,94) y 0,96 (0,93-0,98) para las enfermedades con cuerpos de Lewy y de 0,63 (0,24-0,90) y 0,97(0,93-0,99) para la AMS. No hubo diferencias significativas entre los SAAs de α-sinucleína utilizando piel vs. LCR, pero sí que los resultados fueron significativamente peores en los ensayos que utilizaron mucosa olfatoria (sensibilidad de 0,64 [0,49-0,76]). Posteriormente, se publicó un trabajo incluyendo 1123 participantes (545 con EP, 163 controles sanos, 54 sin déficit dopaminérgico, 51 con síntomas prodrómicos y 310 portadores de mutaciones presintomáticos). Encontraron una sensibilidad de EP respecto a controles de 87,7% (intervalo de confianza del 95% (IC 95): 84,9-90,5), pero, al igual que otros grupos37 (NE-II), una sensibilidad menor en PD LRRK2 positivos 67,5% (IC 95: 59,2-75,8). También se ha demostrado que los SAA proporcionan una elevada capacidad predictiva para el desarrollo de EP o DCL en pacientes con síntomas prodrómicos, aunque en algún estudio esta ha sido moderada13,31,41,42 (NE-II).
A pesar de que los SAA son técnicas prometedoras, existen varios retos y limitaciones que necesitan ser mejorados. En primer lugar, es una técnica difícil de instaurar, ya que pequeñas variaciones en alguno de los parámetros del experimento pueden alterar los resultados y falta estandarizar la metodología entre los diferentes laboratorios. La cuantificación de resultados, aunque abordada en algunos estudios, sigue siendo una necesidad no satisfecha, ya que los resultados siguen siendo en su mayoría dicotómicos (curvas positivas vs. negativas, generalmente con 3 o 4 evaluaciones por muestra si se utiliza LCR o entre 4-8 biopsias por paciente si se emplea tejido cutáneo). Los resultados del SAA de α-sinucleína en piel varían según el número y la localización de las biopsias realizadas. Otro aspecto que debe estudiarse más es la presencia de resultados positivos debido a la existencia de copatología α-sinucleína en enfermedades caracterizadas por la agregación y depósito de otras proteínas43-45 (NE-II). Además, la sensibilidad para la sinucleinopatía AMS ha sido en general decepcionante, alrededor del 20%43,46 (NE-II), aunque algunos estudios han informado cifras con amplificación más tardía y curvas de fluorescencia máxima más bajas que la EP40,42,47. Algunos trabajos para aumentar el rendimiento diagnóstico entre EP y AMS han incorporado otros marcadores además del resultado del SAA de aS como son las cadenas ligeras de neurofilamentos (Nfl), la proteína ácida fibrilar glial (GFAP), o la ubiquitina C-terminal hidrolasa (UCH-L1)48-50 o parámetros radiológicos42,51 (NE-II), pero esto está limitado a la investigación y no hay evidencia suficiente para utilizarlo en la práctica clínica diaria.

13. Siderowf A, Concha-Marambio L, Lafontant DE, et al. Assessment of heterogeneity among participants in the Parkinson’s progression markers initiative cohort using alpha-synuclein seed amplification: a cross-sectional study. Lancet Neurol. 2023;22(5):407-17.
31. Fairfoul G, McGuire LI, Pal S, et al. Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann Clin Transl Neurol. 2016;3(10):812-818.
32. Painous C, Fernández M, Pérez J, et al. Fluid and tissue biomarkers in Parkinson›s disease: Immunodetection or seed amplification? Central or peripheral? Parkinsonism Relat Disord. 2024;121:105968.
33. Höglinger GU, Adler CH, Berg D, et al. A biological classification of Parkinson›s disease: the SynNeurGe research diagnostic criteria. Lancet Neurol. 2024;23(2):191-204.
34. Shahnawaz M, Tokuda T, Waragai M, et al. Development of a biochemical diagnosis of Parkinson disease by detection of α-synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol. 2017;74(2):163-172.
36. Yoo D, Bang JI, Ahn C, et al. Diagnostic value of α-synuclein seeding amplification assays in α-synucleinopathies: A systematic review and meta-analysis. Parkinsonism Relat Disord. 2022 Nov;104:99-109.
37. Garrido A, Fairfoul G, Tolosa E, et al; Barcelona LRRK2 Study Group. α-synuclein RTQuIC in cerebrospinal fluid of LRRK2-linked Parkinson›s disease. Ann Clin Transl Neurol. 2019;6(6):1024-1032.
40. Shahnawaz M, Mukherjee A, Pritzkow S, et al. Discriminating α-synuclein strains in Parkinson› s disease and multiple system atrophy. Nature. 2020 Feb;578(7794):273-277.
41. Iranzo A, Fairfoul G, Ayudhaya ACN, et al. Detection of α-synuclein in CSF by RT-QuIC in patients with isolated rapid-eye-movement sleep behaviour disorder: a longitudinal observational study. Lancet Neurol. 2021 Mar;20(3):203-212.
42. Poggiolini I, Gupta V, Lawton M, et al. Diagnostic value of cerebrospinal fluid alpha-synuclein seed quantification in synucleinopathies. Brain. 2022 Apr 18;145(2):584-595.
43. Compta Y, Painous C, Soto M, et al. Combined CSF α-SYN RT-QuIC, CSF NFL and midbrain- pons planimetry in degenerative parkinsonisms: From bedside to bench, and back again. Parkinsonism Relat Disord. 2022 Jun;99:33-41.
44. Dugger BN, Adler CH, Shill HA, et al; Arizona Parkinson›s Disease Consortium. Concomitant pathologies among a spectrum of parkinsonian disorders. Parkinsonism Relat Disord. 2014 May;20(5):525-9.
45. Robinson JL, Lee EB, Xie SX, et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain. 2018 Jul 1;141(7):2181-2193.
46. Rossi M, Candelise N, Baiardi S, et al. Ultrasensitive RT-QuIC assay with high sensitivity and specificity for Lewy body-associated synucleinopathies. Acta Neuropathol. 2020;140(1):49-62.
47. Ma Y, Farris CM, Weber S, Schade S, Nguyen H, Pérez-Soriano A, et al. Sensitivity and specificity of a seed amplification assay for diagnosis of multiple system atrophy: a multicentre cohort study. Lancet Neurol. 2024 Dec;23(12):1225-1237.
48. Schulz I, Kruse N, Gera RG, Kremer T, Cedarbaum J, Barbour R, et al. Systematic Assessment of 10 Biomarker Candidates Focusing on α-Synuclein-Related Disorders. Mov Disord. 2021 Dec;36(12):2874-2887.
49. Canaslan S, Schmitz M, Villar-Piqué A, Maass F, Gmitterová K, Varges D, et al. Detection of Cerebrospinal Fluid Neurofilament Light Chain as a Marker for Alpha-Synucleinopathies. Front Aging Neurosci. 2021 Sep 22;13:717930.
50. Mondello S, Constantinescu R, Zetterberg H, Andreasson U, Holmberg B, Jeromin A. CSF α-synuclein and UCH-L1 levels in Parkinson’s disease and atypical parkinsonian disorders. Parkinsonism Relat Disord. 2014 Apr;20(4):382-7.
51. Quadalti C, Calandra-Buonaura G, Baiardi S, et al. Neurofilament light chain and α-synuclein RT-QuIC as differential diagnostic biomarkers in parkinsonisms and related syndromes. NPJ Parkinsons Dis. 2021 Oct 11;7(1):93.
Recientemente, dos grupos han intentado definir la EP en función de los procesos patobiológicos subyacentes, proponiendo un sistema de estadificación cada uno de ellos que, con sus diferencias, comparten el considerar diferentes combinaciones de presencia de indicadores biológicos de agregación anómala de α-sinucleína, presencia de alteraciones genéticas asociadas a la EP y la documentación de degeneración dopa- minérgica por imagen52,53 (NE-IV). Históricamente, la EP ha sido definida únicamente por la presentación clínica, pero es sabido que en el momento en que los pacientes manifiestan síntomas motores y son diagnosticados con EP temprana, ya ha tenido lugar una cantidad sustancial de neurodegeneración54 (NE-II). Además, en los últimos años la heterogeneidad de la EP se ha vuelto cada vez más evidente. El objetivo de estos dos trabajos es agrupar a los pacientes por características biológicas para obtener grupos más uniformes y poder realizar un diagnóstico precoz.
La principal ventaja es que una clasificación biológica de la EP probablemente sea imperativa para que los fármacos probados en los ensayos terapéuticos sean eficaces.
El principal inconveniente es el posible impacto negativo ético y social en personas asintomáticas pero que presenten biomarcadores positivos, y que serán etiquetados como enfermas. Además, en algunos aspectos es incierto que se cumplan los requisitos para considerarlas adecuadas para el estadiaje, ya que no queda claro que sean secuenciales y reflejen la progresión de la enfermedad, puesto que los estadios numéricamente inferiores podrían al menos teóricamente no siempre preceder a los superiores55 (NE-IV). Por ello, los autores ratifican que el uso de estas clasificaciones está reservado para investigación en el momento actual.

52. Simuni T, Chahine LM, Poston K, et al. A biological definition of neuronal α-synuclein disease: towards an integrated staging system for research. Lancet Neurol. 2024 Feb;23(2):178-190.
53. Höglinger GU, Adler CH, Berg D, et al. A biological classification of Parkinson›s disease: the SynNeurGe research diagnostic criteria. Lancet Neurol. 2024 Feb;23(2):191-204. Erratum in: Lancet Neurol. 2024 Mar;23(3).
54. Kordower JH, Olanow CW, Dodiya HB, et al. Disease duration and the integrity of the nigrostriatal system in Parkinson›s disease. Brain. 2013 Aug;136(Pt 8):2419-31.
55. Cardoso F, Goetz CG, Mestre TA, et al. A Statement of the MDS on Biological Definition, Staging, and Classification of Parkinson’s disease. Mov Disord. 2024 Feb;39(2):259-266.
56. Obeso JA, Calabressi P. Parkinson’s disease is a recognisable and useful diagnostic entity. Lancet Neurol. 2024 Feb;23(2):133-134.
Los test farmacológicos con agentes dopaminérgicos consisten en la evaluación de los efectos clínicos de la facilitación rápida de la transmisión dopaminérgica central. Uno de los usos que se ha dado a dichas pruebas ha sido el de proporcionar apoyo al diagnóstico clínico de la EP, partiendo de la premisa de que la respuesta de los signos motores a la administración de fármacos dopaminérgicos es un criterio necesario para el diagnóstico de la EP57-60 (NE-II).
Los dos fármacos empleados en este tipo de test son la LD, que en tanto que precursor de la dopamina implica mecanismos presinápticos (síntesis de dopamina a partir de la LD) y postsinápticos (unión al receptor dopaminérgico), y la apormofina, cuya acción es eminentemente postsináptica, al tratarse de un agonista dopaminérgico58,59 (NE-II).
Estos test se pueden emplear tanto en pacientes ambulantes como hospitalizados. Cuando se emplean con fines terapéuticos, habitualmente se realizan en pacientes ya tratados en periodo off, pudiéndose entender este como un off natural o habitual (esto es, el que no es producido por una suspensión o discontinuación de la medicación crónica) o como un off inducido tras la retirada de la medicación por un periodo de horas que teóricamente debiera durar de 3 a 5 semividas del fármaco o los fármacos empleados, pero nunca más de lo que el paciente pueda llegar a tolerar, siendo una norma habitual la de 12 horas de suspensión (comúnmente entre la última toma de la noche de un día y la primera del día siguiente). Por el contrario, cuando se emplean con fines diagnósticos el perfil es diferente (respuesta a fármacos antidopaminérgicos desconocida o incierta, ausencia de fluctuaciones motoras, etc.), por lo que se llevan a cabo en la situación clínica basal o habitual del paciente60-62 (NE-II).
Se lleva a cabo típicamente por la mañana, tras la discontinuación de todas las medicaciones antiparkinsonianas durante la noche previa y habitualmente en situación de ayuno también nocturno. Si bien habitualmente se emplea una formulación oral estándar, la absorción se puede acelerar mediante formulaciones en suspensión (Madopar® dispersable, no disponible en España, o Sinemet® triturado) en 100-150 ml de agua carbonatada. En la última década, la difusión del uso de la infusión continua de gel intestinal de LD ofrece la opción de realizar una prueba nasoduodenal con esta formulación de LD con finalidad de prueba farmacológica para valorar la respuesta o falta de respuesta en pacientes en que se plantee la disyuntiva entre falta de respuesta por problema de absorción (por ejemplo, gastroparesia asociada a la EP) frente a parkinsonismo atípico con falta de respuesta a LD por degeneración postsináptica a nivel nigrostriatal. La reciente introducción de la LD inhalada y subcutánea tras los preceptivos ensayos clínicos fase 3 pivotales y aprobaciones por las correspondientes agencias del medicamento (en nuestro caso la EMA y la AEMPS) abre la posibilidad de su empleo con fines no solo terapéuticos sino también diagnósticos; si bien la evidencia es escasa, la base racional sería la misma, y podría ser interesante en casos en los que se sospecha que la aparente falta de respuesta a la LD oral no se deba a un diagnóstico alternativo sino a la presencia de una mala absorción intestinal del fármaco por vía oral.
En pacientes no tratados previamente se recomienda una dosis de hasta 250 mg, pero en pacientes en tratamiento crónico se suele recurrir a una dosis “supramaximal” (dosis superior a la primera dosis del día; por ejemplo, una dosis 1,5 veces la dosis matutina)63 (NE-II). Se debe esperar una respuesta clínica no antes de los 30 minutos posteriores a la administración de la dosis de LD, observándose el efecto máximo hacia los 45-90 minutos, con una duración que puede ser de varias horas.
Se basa en la inyección subcutánea de este fármaco, ya sea como inyección única de 2 o 3 mg o en forma de dosis repetidas iniciando con 1,5 mg y prosiguiendo con incrementos escalonados de 1,5 a 3 mg a intervalos que se desaconseja sean inferiores a 45 minutos (intervalos inferiores pueden conducir a fracaso de la dosis por hallarse los receptores dopaminérgicos aún ocupados). Se recomienda impregnar al paciente con domperidona por lo menos de 3 a 7 días antes de la prueba, por la frecuencia de reacciones adversas en forma de náuseas e hipotensión ortostática. La respuesta clínica es esperable en torno a los 10 minutos de la inyección, con un efecto máximo hacia los 15-25 minutos y una duración de aproximadamente 60 minutos58 (NE-II).
En comparación con la LD, la apomorfina tiene el atractivo, al menos teórico, de no predisponer a las discinesias, pero, por el contrario, tiene una peor tolerancia, y al producir síntomas muy conspicuos (bostezos) puede fácilmente desenmascarar una evaluación ciega. También puede producir hipotensión ortostática más grave que la LD. Como sucede con las formulaciones no orales de la LD, la apomorfina puede permitir evaluar una respuesta subóptima a la LD hipotéticamente secundaria a una mala absorción intestinal de la LD oral.
Nuevamente, de forma semejante a lo que sucede con la LD, la próxima comercialización de la apomorfina sublingual podría abrir la posibilidad de efectuar el test de apomorfina empleando esta vía en vez de la subcutánea.
Debe ser llevada a cabo por profesionales experimentados, apoyándose idealmente en la videofilmación, que permita la validación a posteriori por parte de otros observadores. Se considera necesaria una mejoría motora de al menos un 30% (habitualmente medida en base a la diferencia entre la sección motora de la escala unificada de la EP, actualmente la MDS-UPDRS, basal y postest)64,65 (NE-II).
La falta de mejoría se ha de tomar con cautela, ya que hasta un 40% de los casos con respuesta negativa a una prueba farmacológica aguda pueden experimentar posteriormente una respuesta adecuada al tratamiento crónico con LD62 (NE-II).
En pacientes de novo esto puede deberse a que se ha empleado una dosis subumbral, por lo que es aconsejable repetir la prueba con aumentos graduales del 25% de la dosis de LD. Por su parte, en pacientes tratados, un problema de absorción puede ser la explicación de la aparente falta de respuesta, tanto aguda como crónica, a la terapia oral. En estos casos puede ser útil efectuar un test de apomorfina, o incluso emplear otra vía o forma de administración de LD, como se ha comentado previamente. Por el contrario, en casos en que se realice un test de apomorfina en primer lugar y este resulte negativo, debe realizarse un test de LD, ya que se ha documentado la respuesta a esta tras un test negativo de apomorfina.
Por último, en pacientes tratados no fluctuantes, pero con indicios de tener algún grado de respuesta a la medicación crónica, el efecto de la prueba aguda con una dosis subumbral puede quedar enmascarado por el efecto crónico del tratamiento habitual, incluso a pesar de haber procedido a la preceptiva discontinuación de medicaciones antiparkinsonianas previa a la realización de la prueba.
Actualmente las guías diagnósticas66-68 no recomiendan los test farmacológicos para el diagnóstico diferencial de la EP, puesto que estos no añaden ningún valor al de la observación de la respuesta del tratamiento crónico con LD ambulatorio y además no están exentas de riesgos o efectos adversos59,69-74 (NE-II). Además, en fases iniciales de la enfermedad, estos test farmacológicos presentan menor rendimiento diagnóstico en comparación con el manejo estándar ambulatorio con LD. Por tanto, la recomendación de estas guías es que estas pruebas agudas no deben emplearse, considerando que los pacientes en los que la bradicinesia o la rigidez no mejoren con una terapia crónica de al menos 600 mg/día de LD durante 6 semanas son “no respondedores a LD” y en estos casos la medicación debería ser suspendida gradualmente.
Estos test pueden ser útiles en otras situaciones, como, por ejemplo, para evaluar la respuesta a la medicación dopaminérgica en personas con EP candidatos a terapias avanzadas o para valorar la falta de respuesta de terapias dopaminérgicas orales secundaria a problemas de absorción (como se ha comentado antes, clásicamente la apomorfina subcutánea, pero actualmente también el gel intestinal de LD y la LD inhalada, la LD subcutánea y en un futuro inmediato la apomorfina sublingual). El test de LD también puede ser de utilidad en la reevaluación de los pacientes cuando tienen problemas relacionados con la respuesta a la medicación, como la latencia o el inicio del beneficio, la magnitud de la respuesta y la duración del beneficio62 (NE-II). Hay pacientes con un diagnóstico de EP idiopática que informan haber experimentado una respuesta subóptima con LD, a pesar de que el médico nota una mejoría en su rendimiento en las escalas de evaluación75 (NE-II). El test de LD es una excelente oportunidad tanto para el médico para confirmar la respuesta a la medicación como para el paciente para darse cuenta de que los síntomas mejoran.

57. Esteguy M, Bonnet AM, Kefalos J, et al. Le test a le levodopa dans la maladie de Parkinson. Rev Neurol (Paris). 1985;141(Suppl):413-4.
58. Barker R, Duncan J, Lees AJ. Subcutaneous apomorphine as a diagnostic test for dopaminergic responsiveness in parkinsonian syndromes. Lancet.1989;1:675.
59. D’Costa DF, Abbott RJ, Pye IF, et al. The apomorphine test in parkinsonian syndromes. J Neurol Neurosurg Psychiatry. 1991;54:870-2.
60. Hughes AJ, Lees AJ, Stern GM. Challenge tests to predict the dopaminergic response in untreated Parkinson’s disease. Neurology. 1991;41:1723-5.
61. Gasser T, Schwarz J, Arnold G, et al. Apomorphine test for dopaminergic responsiveness in patients with previously untreated Parkinson’s disease. Arch Neurol. 1992;49:1131-4.
62. Albanese A, Bonuccelli U, Brefel C, et al. Consensus statement on the role of acute dopaminergic challenge in Parkinson›s disease. Mov Disord. 2001 Mar;16(2):197-201.
63. Merola A, Rizzi L, Zibetti M, et al. Medical therapy and subthalamic deep brain stimulation in advanced Parkinson›s disease: a different long-term outcome? J Neurol Neurosurg Psychiatry. 2014 May;85(5):552-9.
64. Langston JW, Widner H, Goetz CG, et al. Core assessment program for intracerebral transplantations (CAPIT). Mov Disord. 1992;7(1):2-13.
65. Defer GL, Widner H, Marie RM, et al. Core assessment program for surgical interventional therapies in Parkinson’s disease (CAPSIT-PD). Mov Disord. 1999;14:572-84.
66. Parkinson’s disease in adults: diagnosis and management. En: National Institute for Health and Care Excellence (NICE) [Internet]. Disponible en: https://www.nice.org.uk/ guidance/ng71/chapter/Update-information
67. Berardelli A, Wenning GK, Antonini A, et al. EFNS/MDS-ES/ENS [corrected] recommendations for the diagnosis of Parkinson›s disease. Eur J Neurol. 2013 Jan;20(1):16-34.
68. Scottish Intercollegiate Guidelines Network (SIGN). Diagnosis and pharmacological management of Parkinson’s disease. A national clinical guideline. En: Parkinson’s UK [Internet]. Disponible en: https://t.ly/UzriI
69. Clarke CE, Davies P. Systematic review of acute levodopa and apomorphine challenge tests in the diagnosis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2000;69(5):590-594.
70. Rossi P, Colosimo C, Moro E, et al. Acute challenge with apomorphine and levodopa in Parkinsonism. Eur Neurol. 2000;43(2):95-101.
71. Hughes AJ, Lees AJ, Stern GM. Apomorphine test to predict dopaminergic responsiveness in parkinsonian syndromes. Lancet. 1990;336(8706):32-34.
72. Zappia M, Montesanti R, Colao R, et al. Short-term levodopa test assessed by movement time accurately predicts dopaminergic responsiveness in Parkinson’s disease. Mov Disord. 1997 Jan;12(1):103-6.
73. Anderson E, Nutt J. The long-duration response to levodopa: phenomenology, potential mechanisms and clinical implications. Parkinsonism Relat Disord. 2011;17:587-592.
74. Nutt JG, Carter JH, Woodward WR. Long-duration response to levodopa. Neurology. 1995 Aug;45(8):1613-6.
75. Rabel C, Le Goff F, Lefaucheur R, et al. Subjective Perceived Motor Improvement after Acute Levodopa Challenge in Parkinson›s Disease. J Parkinsons Dis. 2016 Oct 19;6(4):779-785.
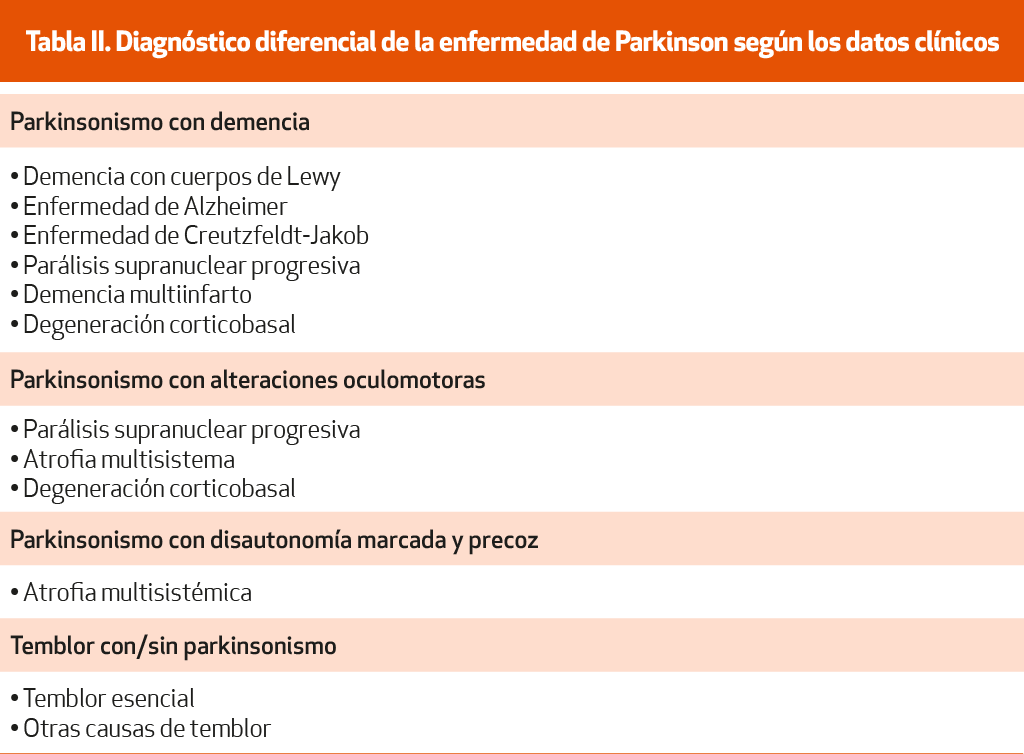
El diagnóstico diferencial de la EP incluye todas las entidades que pueden cursan con temblor o síndrome parkinsoniano, ya sea este un parkinsonismo secundario o un parkinsonismo asociado a otras enfermedades neurodegenerativas diferentes de la EP o a otras causas (Tabla I).

Como se ha comentado previamente, el diagnóstico de la EP es clínico, y es recomendable que el paciente sea remitido sin tratamiento antiparkinsoniano para su evaluación por parte de un especialista en trastornos del movimiento66 (NE-II).
En el diagnóstico diferencial es fundamental comprobar que el paciente no presenta datos clínicos atípicos76 (NE-II), así como que existen datos de apoyo, como un curso progresivo, asimetría y respuesta a la terapia dopaminérgica (ver los criterios diagnósticos de la MDS). La presencia de caídas precoces, la falta de respuesta a la LD, la simetría de los síntomas al inicio, la rápida progresión llegando a un estadio III en la clasificación de Hoehn y Yahr en los 3 primeros años, la ausencia de temblor y la presencia de marcada disautonomía (urgencia o incontinencia urinaria o fecal, retención urinaria, disfunción eréctil o hipotensión ortostática sintomática) son datos clínicos sugestivos de otra causa de parkinsonismo diferente de la EP77 (NE-II). Dado que algunos datos atípicos no estarán presentes al inicio y pueden ir apareciendo a lo largo de la evolución, el diagnóstico debe ser revisado regularmente, reconsiderando siempre la posibilidad de un parkinsonismo atípico en aquellos pacientes que desarrollan signos de evolución atípica66 (NE-IV). La posibilidad de donación del cerebro por parte del paciente podría ser planteada por el clínico para poder hacer el diagnóstico definitivo post mortem66 (NE-IV). La Tabla II muestra diferentes entidades que se han de considerar en el diagnóstico diferencial de la EP teniendo en cuenta algunos aspectos clínicos.
No sería preciso realizar un estudio de neuroimagen estructural (tomografía computarizada [TC] o RM craneal) de forma rutinaria a todo paciente con sospecha de EP78 (NE-III). Sin embargo, sí sería recomendable realizar una TC craneal a todo paciente con sospecha de patología cerebral (tumor, hidrocefalia, etc.) que pueda estar causando o agravando el síndrome parkinsoniano68 (NE-IV), así como una RM craneal en caso de sospecha de enfermedad vascular cerebral subcortical causante de un parkinsonismo vascular o datos atípicos que sugieren un parkinsonismo atípico68 (NE-IV). Para revisar en profundidad la utilidad de las técnicas de imagen en el diagnóstico y diagnóstico diferencial de la EP, remitimos al lector al capítulo 2, donde se trata este tema de manera específica (pag. 57).
Además, en un paciente con sospecha de EP es recomendable una evaluación neuropsicológica que incluya una entrevista con el cuidador del paciente, evaluación de funciones cognitivas y cribado para trastorno de conducta del sueño REM, síntomas psicóticos y depresión67 (NE-I). No será necesario realizar estudios analíticos en el caso de una EP típica, pero serán útiles y necesarios determinados estudios (hormonas tiroideas, niveles séricos de ceruloplasmina, cobre en orina y suero, acantocitos en sangre periférica, creatina cinasa, estudio del hierro, estudios genéticos, etc.) en aquellos pacientes en los que los síntomas no estén tan claros y en los que se deban excluir otras entidades.

Los parkinsonismos secundarios o sintomáticos son aquellos síndromes parkinsonianos producidos por una causa conocida y no por una enfermedad degenerativa. Representan entre el 25 y el 50% del total de los síndromes parkinsonianos.
Dentro de ellos, el parkinsonismo inducido por fármacos es el más frecuente. Los factores de riesgo para su desarrollo son la edad avanzada, el sexo femenino, una mayor potencia antidopaminérgica del fármaco, mayor dosis y tiempo de exposición y la presencia de un temblor de acción previo a la administración del fármaco. Será fundamental la prevención, evitando la administración de fármacos antidopaminérgicos, y en el caso de ser preciso su uso, intentar usar los que tienen un menor efecto parkinsonizante, administrarlos durante el menor tiempo y a la menor dosis posible. El parkinsonismo suele ser simétrico (aunque puede ser asimétrico) y asociar discinesias, que pueden desarrollarse tras 3 meses con el tratamiento responsable. En general, el parkinsonismo es reversible tras la supresión del fármaco causal y no se recomienda el uso de anticolinérgicos. En caso de no resolución después de 6 meses de haber retirado el fármaco, nos deberemos plantear si el paciente podría presentar una EP latente precipitada por el mismo, para lo que puede resultar útil realizar una prueba de neuroimagen funcional de la vía nigroestriada presináptica como una SPECT 3I-β-CIT (DaTSCAN®), que será normal en el caso de un parkinsonismo inducido por fármacos68,77 (NE-II).
En otras ocasiones, el parkinsonismo es ocasionado por tóxicos que dañan estructural y funcionalmente los ganglios basales, resultando muchas veces los síntomas irreversibles.
La Tabla III muestra los principales fármacos y tóxicos causantes de parkinsonismo.
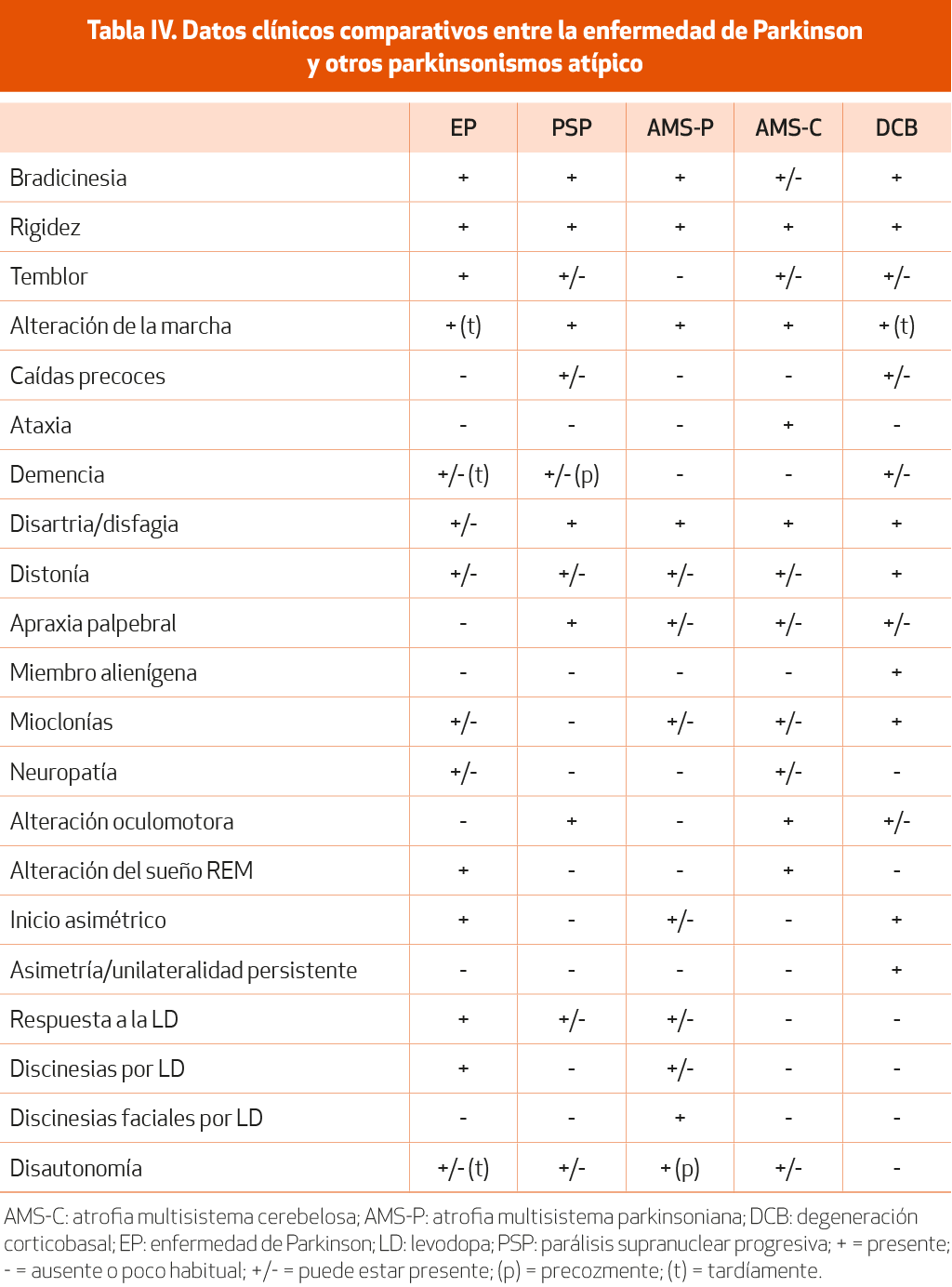
Los pacientes con parkinsonismos atípico (AMS, PSP y degeneración corticobasal [DCB]) plantean en ocasiones un reto diagnóstico, sobre todo en etapas tempranas de la enfermedad. Solo del 65 al 77% de los casos diagnosticados a finales de los años ochenta de EP idiopática cumplían criterios anatomopatológicos de EP1,3 (NE-II), el resto correspondían a otras enfermedades degenerativas que comprometían la vía nigroestriatal. Lo característico de los parkinsonismos atípicos es la nula o escasa y temporal respuesta a la LD y un peor pronóstico con una esperanza de vida reducida. En 2017 se publicaron los nuevos criterios de la PSP79 (NE-II) que, a diferencia de los previos, incluyen otras variantes además de la PSP richardsoniana. La variante PSP-parkinsonismo en concreto es la que más se asemeja a la EP, ya que los pacientes pueden presentar respuesta a la LD (habitualmente de forma temporal y baja-moderada) y un parkinsonismo simétrico o asimétrico que puede asociar temblor de reposo, que por tanto ha dejado de ser criterio de exclusión de PSP. La presencia de alteraciones oculomotoras en el plano vertical (enlentecimiento de los movimientos sacádicos o limitación de la infraversión de la mirada) sugieren el diagnóstico de PSP, pero pueden no estar presentes al inicio o incluso no estarlo en todo el transcurso de la enfermedad80 (NE-III). En cuanto a la AMS, los nuevos criterios se publicaron en 202281 (NE-II) e incluyen cuatro grados de certeza diagnóstica (definitiva, clínicamente definida, probable y posible prodrómica) y los dos fenotipos previamente conocidos (AMS-parkinsonismo y cerebelosa).
Los datos clínicos básicos comparativos vienen reflejados en la Tabla IV. Para cada uno de ellos existen criterios de certeza, y en todos los casos es necesaria la anatomía patológica para el diagnóstico definitivo.
En el capítulo 2 se revisa la utilidad de las pruebas de imagen en el diagnóstico de la EP y los parkinsonismos atípicos (pag. 57). Pruebas de olfacción como el Brief Smell Identification Test (BSIT), el University of Pennsylvania Smell Identification Test (UPSIT) o el San Diego Odor Identification Test (SDOIT) no se recomiendan en el diagnóstico diferencial de la EP con los otros parkinsonismos neurodegenerativos, porque no hay suficiente evidencia que lo apoye66 (NE-II). Los estudios de función autonómica (cardiovascular, urinaria, anorrectal y temperatura cutánea y sudoración) tampoco han demostrado ser beneficiosos67,77 (NE-IV).

Por último, el uso de SAA de α-sinucleína en LCR de forma aislada o en combinación con otros biomarcadores, como los niveles en LCR o en sangre de neurofilamentos o medidas de atrofia de mesencéfalo y protuberancia, se está mostrando de forma creciente en publicaciones como sensible y específico para diferenciar los parkinsonismos atípicos de la EP, si bien aún no están disponibles de forma ubicua para poder emplearse en la rutina clínica31,41,43,51 (NE-III). Un ejemplo de situación donde el SAA de α-sinucleína podría ser útil sería la diferenciación entre EP y PSP-P, aunque aún se dispone de evidencia limitada y la presencia de copatología podría en algunos casos justificar un SAA+, aun tratándose de una PSP; en este contexto, todavía es necesaria una metodología perfeccionada de SAA para proteína 4R-τ82 (NE-III).
Otras enfermedades neurodegenerativas que pueden cursar con parkinsonismo y que hay que tener en cuenta en el diagnóstico diferencial se reflejan en la Tabla II.
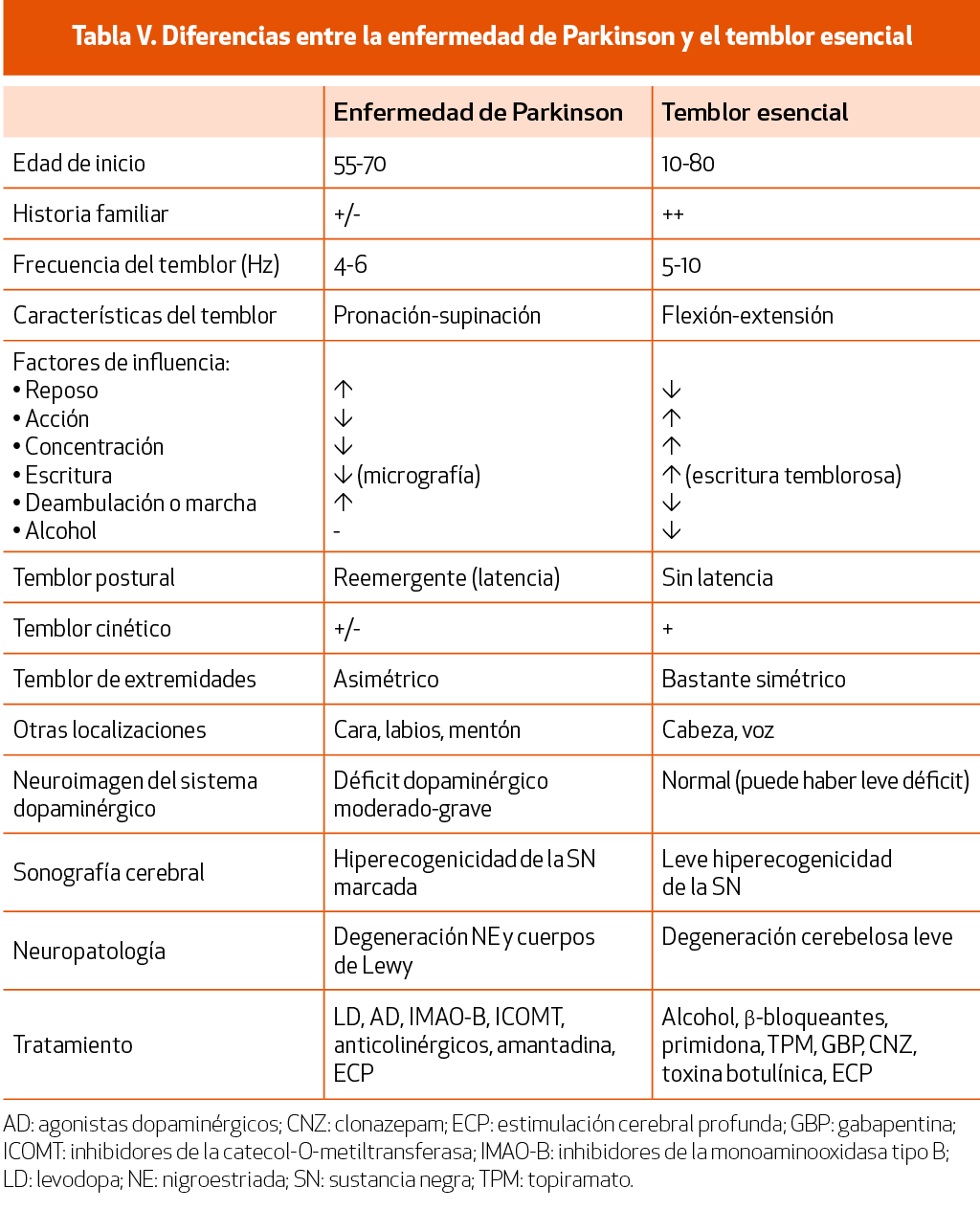
El temblor esencial (TE) es, posiblemente, la entidad con la que más frecuentemente se plantea el diagnóstico diferencial de la EP. Lo característico del TE (Tabla V) será la presencia de antecedentes familiares positivos de temblor, fundamentalmente postural de manos bilateral y relativamente simétrico, de cabeza y voz, y mejoría del temblor con el consumo de alcohol; no esperaríamos encontrar otros datos de parkinsonismo, aunque en ocasiones puede asociar temblor de reposo. En casos de duda, un DaTSCAN® podría ser de ayuda en el diagnóstico diferencial con la EP, que será normal en caso de TE67 (NICE 2006) (NE-III). Algunos estudios han observado que los pacientes con TE tienen más probabilidad de desarrollar EP83, por lo que se deberá tener en cuenta en su seguimiento evolutivo. El SAA de α-sinucleína negativo también puede añadirse al arsenal diagnóstico diferencial43 (NE-IV).
El parkinsonismo funcional es una causa poco frecuente. En ocasiones puede coexistir un parkinsonismo o EP con componente funcional asociado. La utilización combinada de la clínica, electrofisiología y neuroimagen funcional68,77,84 (NE-II) mejora la fiabilidad diagnóstica para distinguir entre formas de parkinsonismo funcional puras y combinadas. El parkinsonismo vascular típicamente es de predominio en extremidades inferiores, hay poca respuesta a la LD y daño vascular subcortical en los estudios de imagen craneal. Otras patologías que pueden simular un síndrome parkinsoniano y deben ser consideradas en el diagnóstico diferencial de la EP son por ejemplo un hipotiroidismo, la depresión o la patología osteoarticular.

1. Rajput AH, Rozdilsky B, Rajput A. Accuracy of clinical diagnosis in parkinsonism: a prospective study. Can J Neurol Sci. 1991;18:275-8.
3. Hughes AJ, Daniel SE, Kilford L, et al. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55:181-4. d
31. Fairfoul G, McGuire LI, Pal S, et al. Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann Clin Transl Neurol. 2016;3(10):812-818.
41. Iranzo A, Fairfoul G, Ayudhaya ACN, et al. Detection of α-synuclein in CSF by RT-QuIC in patients with isolated rapid-eye-movement sleep behaviour disorder: a longitudinal observational study. Lancet Neurol. 2021 Mar;20(3):203-212.
43. Compta Y, Painous C, Soto M, et al. Combined CSF α-SYN RT-QuIC, CSF NFL and midbrain-pons planimetry in degenerative parkinsonisms: From bedside to bench, and back again. Parkinsonism Relat Disord. 2022 Jun;99:33-41.
51. Quadalti C, Calandra-Buonaura G, Baiardi S, et al. Neurofilament light chain and α-synuclein RT-QuIC as differential diagnostic biomarkers in parkinsonisms and related syndromes. NPJ Parkinsons Dis. 2021 Oct 11;7(1):93.
66. Parkinson’s disease in adults: diagnosis and management. En: National Institute for Health and Care Excellence (NICE) [Internet]. Disponible en: https://www.nice.org.uk/ guidance/ng71/chapter/Update-information
67. Berardelli A, Wenning GK, Antonini A, et al. EFNS/MDS-ES/ENS [corrected] recommendations for the diagnosis of Parkinson›s disease. Eur J Neurol. 2013 Jan;20(1):16-34.
68. Scottish Intercollegiate Guidelines Network (SIGN). Diagnosis and pharmacological management of Parkinson’s disease. A national clinical guideline. En: Parkinson’s UK [Internet]. Disponible en: https://t.ly/UzriI
76. Gibb WR, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1988;51:745-52.
77. Suchowersky O, Reich S, Perlmutter J, et al.; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: diagnosis and prognosis of new onset Parkinson disease (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2006;66(7):968-75.
78. NHS Quality Improvement Scotland. Diagnosis and pharmacological management of Parkinson’s disease. A national clinical guideline. Edimburgo: Scottish Intercollegiate Guidelines Network, EH7 5EA; 2010.
79. Höglinger GU, Respondek G, Stamelou M, et al.; Movement Disorder Society-endorsed PSP Study Group. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov Disord. 2017 Jun;32(6):853-864.
80. Williams DR, de Silva R, Paviour DC, et al. Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson›s syndrome and PSP-parkinsonism. Brain. 2005 Jun;128(Pt 6):124.
81. Wenning GK, Stankovic I, Vignatelli L, et al. The Movement Disorder Society Criteria for the Diagnosis of Multiple System Atrophy. Mov Disord. 2022 Jun;37(6):1131-1148. doi: 10.1002/mds.29005.
82. Saijo E, Metrick MA 2nd, Koga S, et al. 4-Repeat tau seeds and templating subtypes as brain and CSF biomarkers of frontotemporal lobar degeneration. Acta Neuropathol. 2020 Jan;139(1):63-77.
83. Benito-León J, Louis ED, Bermejo-Pareja F; Neurological Disorders in Central Spain Study Group. Risk of incident Parkinson’s disease and parkinsonism in essential tremor: a population based study. J Neurol Neurosurg Psychiatry. 2009;80(4):423-5.
84. Grimes D, Gordon J, Snelgrove B, et al. Canadian Guidelines on Parkinson’s disease. Can J Neurol Sci. 2012 Jul;39(4 Suppl 4):S1-30.