

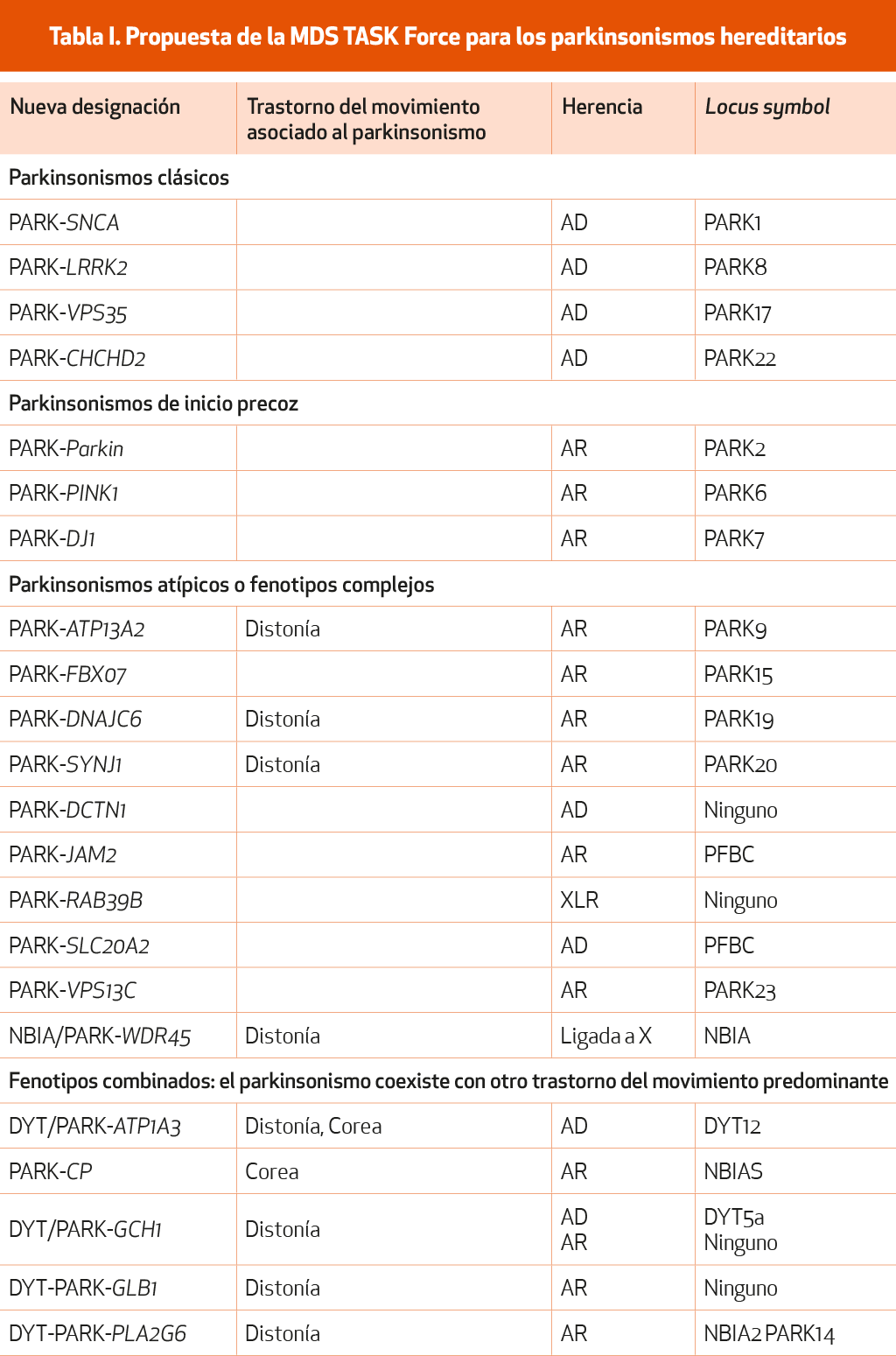
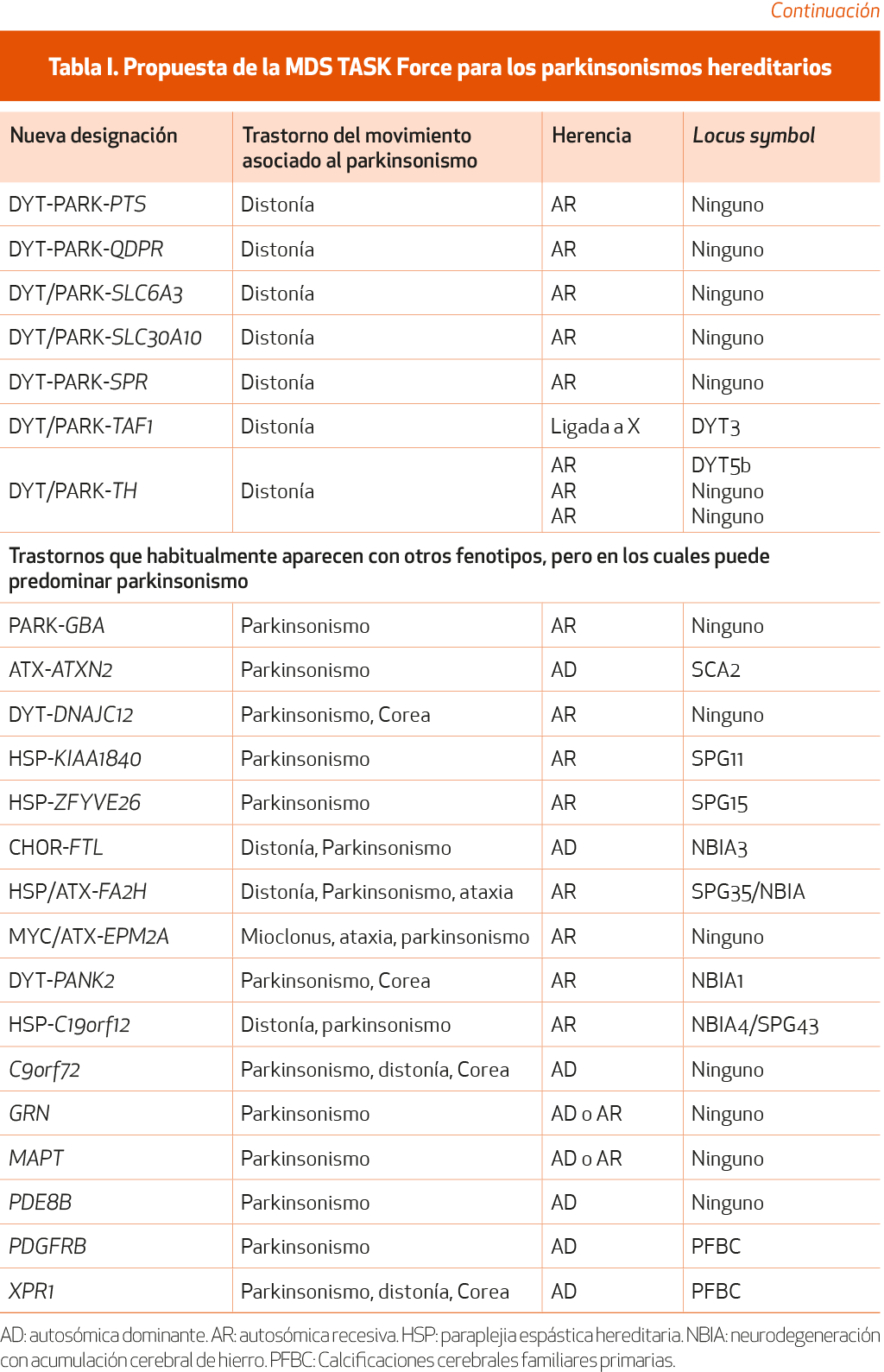
Han pasado más de 25 años desde la descripción de la primera mutación en el gen α-sinucleína (SNCA) asociada a la EP. Desde entonces se han identificado mutaciones en más de 20 genes distintos asociados a la EP. De manera global, estas mutaciones podrían explicar cerca del 30 % de las formas familiares de EP y el 3-5 % de las formas esporádicas. El sistema de locus asignados a símbolos (DYT, PARK, etc.) utilizado hasta recientemente se estableció originalmente para especificar regiones cromosómicas que habían sido ligadas a un trastorno familiar, aunque el gen fuera desconocido. Este sistema, más fácil de recordar, se aceptó por los clínicos, pero el avance genético planteó problemas como relaciones inconsistentes entre los miembros afectos y el fenotipo de los trastornos de movimiento, la existencia de más de un símbolo asignado para el mismo trastorno o incluso el hecho de que no se hayan confirmado asociaciones entre un gen o locus y un trastorno del movimiento. En la actualidad se han asignado 23 genes y locus al acrónimo PARK. En los casos previamente denominados como PARK 3, 5, 11, 13, 18 y 21, la relación no está confirmada. Además, las tres entidades previamente conocidas como PARK 10, 12 y 16 han pasado a la categoría de factor de riesgo. Finalmente, el PARK 1 y el PARK 4 se refieren al gen SNCA y son idénticos. Considerando los avances habidos en genética, que justifican un posible componente hereditario en esta enfermedad, el Task Force on Genetic Nomenclature in Movement Disorders de la MSD publicó en 2016 unas recomendaciones en las que se revisaba la nomenclatura de la genética de los trastornos de movimiento1 (NE-III) y se consideraban las condiciones necesarias para garantizar la evidencia imprescindible al asociar una alteración genética a una enfermedad determinada. Basándose en estas recomendaciones, el MSD Task Force presentó una nueva lista de parkinsonismos hereditarios, que ha sido actualizada recientemente2 (NE-III) (Tabla I).
1. Marras C, Lang A, Van de Warrenburg BP, et al. Nomenclature of Genetic Movement Disorders: Recommendations of the International Parkinson and Movement Disorder Society Task Force. Mov Disord. 2016;31:436-57.
2. Lange LM, González-Latapi P, Rajalingam R, et al; on behalf of the Task Force on Genetic Nomenclature in Movement Disorders. Nomenclature of Genetic Movement Disorders: Recommendations of the International Parkinson and Movement Disorder Society Task Force - An Update. Mov Disord. 2022 May;37(5):905-935.
Las recomendaciones del grupo de trabajo son las siguientes:
Incluir solo aquellos genes donde es posible realizar una prueba genética diagnóstica.
Asignar un prefijo al fenotipo que lo relacione con el mismo. Por ejemplo, cuando el parkinsonismo es lo más llamativo de la clínica, se le asignaría el prefijo PARK, pero si es la distonía el síntoma predominante, se asignaría el prefijo DYT/PARK, como por ejemplo en el caso de DYT/PARK-ATP1A3.
Reemplazar el número del sufijo por el nombre del Por ejemplo, no se empleará en lo sucesivo PARK8, sino PARK-LRRK2 (Tabla I).
Separar las listas de los genes causantes de enfermedad de aquellos genes que constituyen un factor de riesgo. Sería el caso de las mutaciones en heterocigosis en el gen de la glucocerebrosidasa (GBA), que no se consideran una causa monogénica de parkinsonismo porque en sí mismas solo suponen un factor de riesgo para padecer EP, especialmente entre los judíos askenazíes, y que aparece en algunos casos con baja penetrancia.
Elegir el umbral de evidencia antes de asignar un locus symbol. El cumplimiento de este punto conlleva el nivel de evidencia de la causalidad en una EP de origen hereditario, y exige:
Presencia de las variantes patogénicas en múltiples individuos afectados no relacionados.
Evidencia para la segregación o asociación estadística de una variante con la enfermedad.
La variante debe conservarse a través de diferentes especies.
La variante debería alterar el efecto bioquímico normal de un producto génico y demostrarlo en pruebas funcionales en el tejido humano ya sea celular, en modelos animales, anomalías bioquímicas o histológicas.
Por tanto, la irrupción de la genética nos ha permitido tener una visión distinta de la enfermedad y aproximarnos a nuevos mecanismos patogénicos. Se ha demostrado que numerosos genes causantes de las formas mendelianas de EP, además de justificar el componente familiar de un número importante de casos, podrían tener un rol principal también en la EP esporádica. Este es el caso de algunas mutaciones de las formas monogénicas (por ejemplo, mutaciones en LRRK2) o de los alelos de susceptibilidad (por ejemplo, mutaciones en el gen GBA). Los resultados del último metaanálisis de estudios de asociación genómica han identificado más de 90 loci asociados a un mayor riesgo de desarrollar EP3 (NE-III). Se podría hablar, por tanto, de un espectro de la enfermedad que va desde mutaciones altamente patógenas, pasando por variantes de riesgo moderado, hasta múltiples loci que confieren pequeños incrementos del riesgo de sufrir la enfermedad.
A pesar de que la contribución de la genética a la EP podría ser mayor de lo pensado, seguimos sin conocer la etiopatogenia última de la EP, una enfermedad con un origen probablemente multifactorial y una compleja interrelación de factores genéticos y ambientales, interacciones gen-ambiente (influencia de agentes ambientales en la expresión génica) y su impacto directo en el desarrollo y envejecimiento del cerebro.
Hasta la fecha no existen directrices formalmente aceptadas por las principales sociedades científicas acerca del uso de los test genéticos en el diagnóstico de la EP, aunque, no obstante, se va perfilando una forma habitual de actuación, basada principalmente en las recomendaciones de los grupos de expertos4,5 (NE-IV). La tendencia general es hacia un uso creciente de dichos test genéticos, fruto por un lado de un mejor acceso a los mismos y por otro de la llegada de los primeros ensayos clínicos con terapias dirigidas a subtipos genéticos específicos (como LRRK2 o GBA).
3. Nalls MA, Blauwendraat C, Vallerga CL, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18:1091-1102.
4. Pal G, Cook L, Schulze J, et al. Genetic Testing in Parkinson’s Disease. Mov Disord. 2023 Aug;38(8):1384-1396.
5. Jesús Mastre S, Santos García D. Recomendaciones para el abordaje de estudios genéticos en trastornos del movimiento, ataxias y paraparesias. Madrid: SEN; 2023.
En la práctica clínica habitual, el empleo del estudio genético con fines diagnósticos está limitado por la reducida proporción de casos de EP en los que se puede encontrar una mutación genética causal (menos del 10-30%, según las poblaciones). Desde una perspectiva clínica, la ausencia de terapias modificadoras de la enfermedad y la penetrancia variable de la mayoría de las variantes conocidas limitan la utilidad de las pruebas de diagnóstico genético para la EP.
En la actualidad, estos estudios se realizan clínicamente en entornos concretos, seleccionando caso a caso. Desde una perspectiva de investigación, el estudio genético se realiza para ayudar en la tipificación, y ofrecer la oportunidad de participar en ensayos clínicos basados en su estatus genético. En este sentido, es probable que en los próximos años vean la luz los primeros fármacos diseñados para formas monogénicas (LRRK2) o variantes de riesgo (GBA), con un potencial beneficio tanto para las formas genéticas como esporádicas de EP.
Las pruebas genéticas pueden proporcionar información útil para los pacientes y sus cuidadores sobre la propia enfermedad, ayudando en la incertidumbre del pronóstico y, en algunos casos, para abordar una planificación reproductiva. Cuando se analiza la opinión de los pacientes, los que han tenido una enfermedad de inicio temprano están de acuerdo en realizarse el estudio genético, como una forma de entender mejor su enfermedad y de tomar decisiones para su futuro4 (NE-IV).
Al propio médico conocer el estatus genético del paciente le puede ofrecer una información adicional que posibilita una mejor orientación terapéutica, derivada de la seguridad del diagnóstico, y valorar la utilidad o no de algunas opciones de tratamiento, como se ha planteado en la cirugía mediante estimulación cerebral profunda en pacientes con variantes en el gen GBA, donde los resultados esperables son peores6.
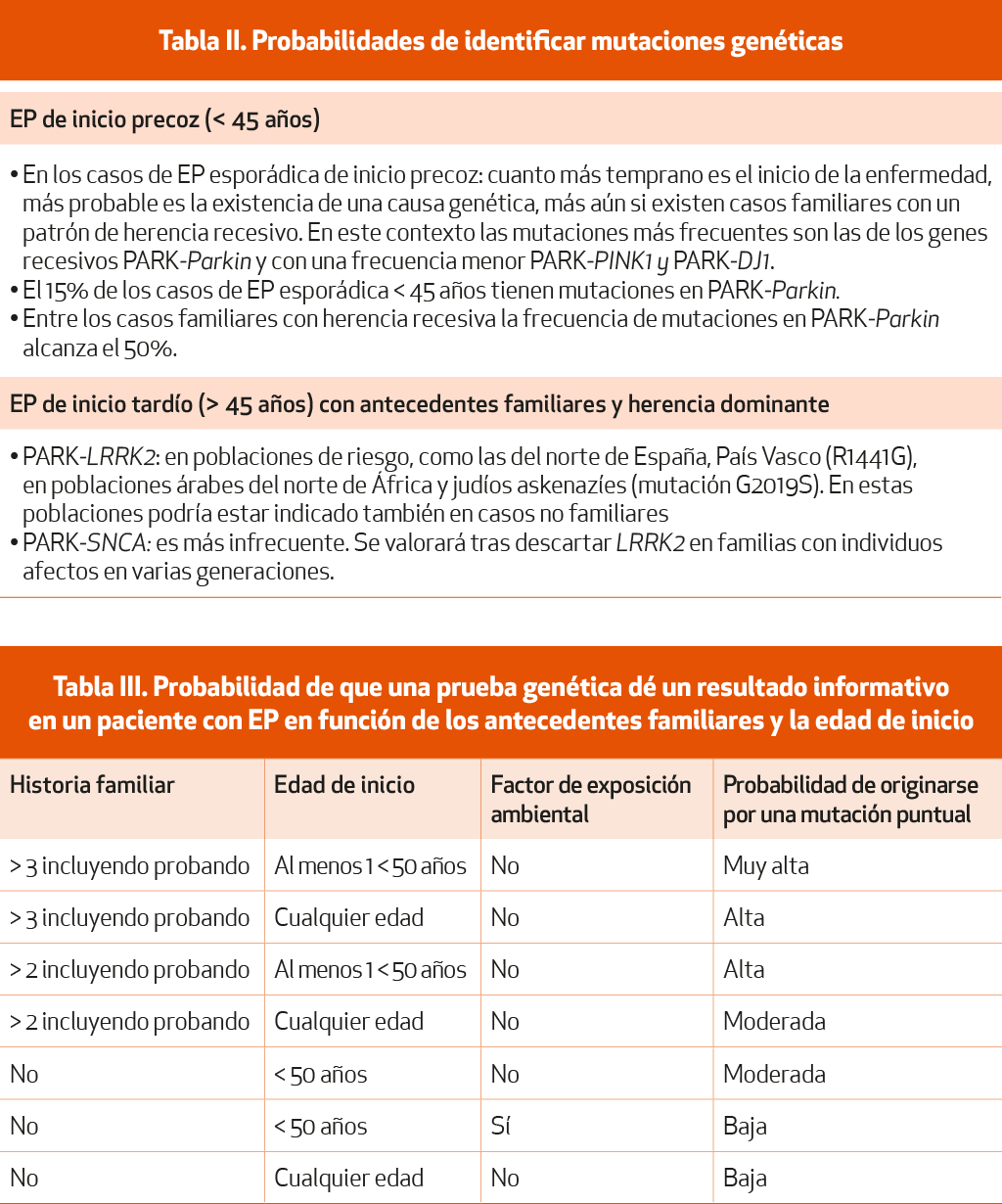
Sin considerar los posibles fármacos específicos actualmente en fase de ensayo, con una perspectiva de coste-eficacia y en el ámbito puramente asistencial, las pruebas genéticas podrían quedar restringidas a aquellas poblaciones en las que es más probable identificar pacientes con EP asociada a una determinada mutación, teniendo en cuenta consideraciones como la edad, el patrón de herencia o los factores raciales (Tablas II y III)7 (NE-III). No obstante, conviene recordar que un estudio negativo no excluye una causa genética, en tanto en cuanto muchas mutaciones genéticas aún se desconocen o solo se han realizado en los genotipos más frecuentes8 (NE-IV).
Tanto en un ámbito asistencial como de investigación, el estudio genético se debe realizar en centros especializados. Se debe informar al paciente y sus familiares con detalle, obtener un consentimiento informado firmado y realizar un asesoramiento genético antes de realizar la prueba y tras el resultado de la misma. La confidencialidad del resultado debe garantizarse, y en todo caso siempre debemos ponderar detenidamente el beneficio que la prueba pueda proporcionar al paciente, más allá de satisfacer la curiosidad del médico que lo trata.
Las probabilidades de identificar mutaciones genéticas causales en pacientes con EP aumentan en dos circunstancias:
EP de inicio precoz (menores de 45 años) y cuando existen antecedentes familiares de la enfermedad8 (NE-IV). En los casos esporádicos de inicio precoz, cuanto más temprano es este inicio, más probable es la existencia de una causa genética, más aún si existen casos familiares con un patrón de herencia recesivo. En este contexto las mutaciones más frecuentes son las de los genes recesivos PARK-Parkin y, con una frecuencia menor, PARK-PINK1 y PARK-DJ1. El 15% de los casos de EP menores de 45 años tienen mutaciones en PARK-Parkin. Entre los casos familiares con herencia recesiva la frecuencia de mutaciones en PARK-Parkin alcanza el 50%.
EP de inicio tardío (mayores de 45 años). En estos casos estará indicada la realización de una prueba genética solo cuando exista una historia familiar de EP con un patrón de herencia autosómica dominante. En estos casos estaría indicado estudiar las mutaciones causales del gen LRRK2, particularmente la G2019S. En determinadas poblaciones en las que se conoce la existencia de una elevada frecuencia de mutaciones en el gen LRRK2, como sucede en el norte de España, y especialmente en el País Vasco (mutación R1441G), en poblaciones árabes del norte de África y en judíos askenazíes (mutación G2019S). En las formas de inicio tardío con individuos afectos en múltiples generaciones, una vez descartadas mutaciones en LRRK2, procede estudiar el gen SNCA, aunque sus mutaciones son muy poco frecuentes incluso en este contexto. A la hora de efectuar el consejo genético en los portadores de mutaciones en LRRK2 debe tenerse en cuenta la penetrancia incompleta de algunas de las mutaciones, así como la patogenicidad dudosa de algunas variantes9 (NE-IV).
Con respecto al tipo de estudio, el tamaño de los paneles multigénicos de laboratorios comerciales es muy variable, con paneles que incluyen desde 5 hasta más de 50 genes. Existe, por lo tanto, también la necesidad de consensuar qué genes deben incluirse en un panel multigénico de laboratorio comercial para el estudio genético de EP10 (NE-IV), aunque la tendencia actual se dirige cada vez más a realizar estudios de secuenciación masiva.
4. Pal G, Cook L, Schulze J, et al. Genetic Testing in Parkinson’s Disease. Mov Disord. 2023 Aug;38(8):1384-1396.
6. Pal G, Mangone G, Hill EJ, et al. Parkinson disease and subthalamic nucleus deep brain stimulation: cognitive effects in GBA mutation carriers. Ann Neurol. 2022;91(3):424-435.
7. Berardelli A, Wenning GK, Antonini A, et al. EFNS/MDS-ES/ENS recommendations for the diagnosis of Parkinson’s disease. Eur J Neurol. 2013;20:16-34.
8. Jacobs H, Latza U, Vieregge A, et al. Attitudes of young patients with Parkinson’s disease towards possible presymptomatic and prenatal genetic testing. Genet Couns. 2001;12:55-67.
9. Klein C, Schlossmacher MG. The genetics of Parkinson disease: Implications for neurological care. Nat Clin Pract Neurol. 2006;2:136-46.
10. Cook L, Schulze J, Verbrugge J, et al.; ClinGen Parkinson’s Disease Gene Curation Expert Panel and the MDS Task Force for Recommendations for Genetic Testing in Parkinson’s Disease; Clinical Genome Resource (ClinGen) Parkinson’s Disease Gene Curation Expert Panel Authors; Movement Society Disorder (MDS) Task Force on Recommendations for Clinical Genetic Testing in Parkinson’s Disease Authors. The commercial genetic testing landscape for Parkinson’s disease. Parkinsonism Relat Disord. 2021 Nov;92:107-111.
La European Federation of Neurological Societies (EFNS) y MDS establecieron en 2013 una serie de recomendaciones, que se han actualizado posteriormente, según las cuales estaría indicada la realización de estudios genéticos para mutaciones específicas en11 (NE-III, grado de recomendación B):
Pacientes con EP típica e historia familiar positiva sugestiva de herencia dominante (gen LRRK2, mutaciones patogénicas).
Pacientes con EP esporádica de inicio tardío, solo en poblaciones con una frecuencia conocida elevada de mutaciones patogénicas (LRRK2-R1441G en País Vasco, LRRK2- G2019S y GBA en judíos askenazíes, LRRK2-G2019S en árabes del norte de África).
Familias con múltiples individuos afectos en más de una generación, sugestivo de herencia dominante, con edad de inicio precoz o tardía (gen SNCA).
EP típica con historia familiar compatible con herencia recesiva, particularmente si la edad de inicio es inferior a 50 años (genes Parkina, PINK1, DJ1).
EP esporádica de inicio precoz, particularmente si el inicio es antes de los 40 años y hay signos atípicos como distonía al inicio (genes Parkina, PINK1, DJ1).
La Sociedad Española de Neurología (SEN), viendo la importancia creciente de esta situación publicó en 2023 el manual denominado Recomendaciones para el abordaje de estudios genéticos en trastornos del movimiento, ataxias y paraparesias5, en el que se sintetizan las recomendaciones más actuales tanto de la EFNS como de la MDS para cada caso, teniendo en cuenta principalmente la edad y los antecedentes familiares, y estableciendo una serie de niveles diagnósticos (NE-IV, grado de recomendación D, también denominado RecSEN en el trabajo referido):
EP de inicio juvenil (< 21 años) y EP de inicio precoz (edad de inicio 21-45 años) independientemente de la presencia de antecedentes familiares.
− Primer nivel: PRKN.
− Segundo nivel: PINK1 y DJ1.
- Tercer nivel: solo en los casos de EP de inicio muy precoz, si no se han detectado mutaciones en los genes PRKN, PINK1 y DJ1, se debería considerar solicitar el genotipado de los genes ATP13A2, PLA2G6 y FBXO7.
- Cuarto nivel: Panel multigénico/WES.
EP de inicio tardío (edad de inicio > 45 años).
− Si tiene antecedentes familiares con un patrón de herencia dominante:
- Primer nivel: se recomienda realizar el estudio de las mutaciones reconocidas como patógenas en el gen LRRK2. En población general debe realizarse el estudio de la mutación G2019S, mientras que en población de ascendencia vasca debe considerarse el estudio de la mutación R1441G.
- Segundo nivel: las mutaciones en SNCA son menos frecuentes, por lo que debe valorarse su estudio tras haber descartado mutaciones en el gen LRRK2 en familias con individuos afectos en varias generaciones.
- Tercer nivel: panel multigénico/WES.
- En judíos askenazíes sin antecedentes familiares también podría estar justificado estudiar las mutaciones G2019S y R1441G, dada su alta prevalencia en esta población.

5. Jesús Mastre S, Santos García D. Recomendaciones para el abordaje de estudios genéticos en trastornos del movimiento, ataxias y paraparesias. Madrid: SEN; 2023.
11. Buhat DM, Tan EK. Genetic testing of LRRK2 in Parkinson’s disease: is there a clinical role? Parkinsonism Relat Disord 2014;20 Suppl 1:S54-6.